Markdown Hosing - Register Now!
Home > BioSeq Utilities | Japanese
BioSeq utilities written in Perl can be used to decode biological sequences (bio-sequence in short) such as nucleotides in a nucleic acid (RNA or DNA) or amino acids representing a protein.
In most cases, a bio-sequence is represented in text format for ease of sharing. UNIX (or Linux) text tools are conveniently used to handle these sequence data. The bioseq utilities are meant to augment Linux command-line tools to assist bio-sequence-specific decoding.
Kobu.Com is good at text processing (search, modification and format conversion) especially when a large amount of documents are batch-processed. Bioinformatics requires skills in information processing as well as expertise in medicine and biology. If you are working in medicine and biology and not very confident in computing, we are ready to help you. We can help you in preparing inspection data for further analysis or submitting the data to a public online database. We can design, develop or customize tools for decoding, modifying and format-converting bio-sequences. Please feel free to contact us.
Currently the following three utilities exist (in written order):
| Utility | Name | Description |
|---|---|---|
| motifgrep | pattern matcher | searches a bio-sequence for occurences that satisfy a motif pattern |
| nucleamino | nucleotide to amino acid converter | converts a nucleotide sequence to an amino acid sequence |
| modshunt | sequence diff | compares two bio-sequences and print differences |
The BioSeq utilities are published on GitHub. See README.md on GitHub for how to setup and use the tools.
The rest of this page introduces the bioseq utilities using examples.
First I describe the test data used in the rest of the page. Two types of bio-sequences of the novel coronavirus (SARS-CoV-2) are used to show how the utilities handle them:
They are downloaded from the NCBI online database run by NIH and others:
Sequences of two viruses are used. One is collected in Wuhan in December 2019 which is used as a reference sequence compared to variant viruses. The other is a variant virus collected in Australia in June 2020. The latter contains 'N501Y' variation said to increase infection power. For each virus, two sets of data, RNA nucleotide sequence and amino acid sequence of the spike protein, are downloaded (in Jan 2021).
| Virus | Sequence Type | Accession | File | S Protein Range |
|---|---|---|---|---|
| RefSeq | Nucleotide | NC_045512.2 | nuc-refseq.fasta | 21563..25384 |
| RefSeq | Surface Glycoprotein | YP_009724390.1 | pro-refseq.fasta | |
| Au N501Y | Nucleotide | MT745734.1 | nuc-au-200617.fasta | 21525..25346 |
| Au N501Y | Surface Glycoprotein | QLG76817.1 | pro-au-200617.fasta |
You can download the same data from the NCBI site by specifying the accession number of the sequence.
A FASTA format is commonly used in online bio-sequence databases such as NCBI. A FASTA file starts with a header line prefixed with '>' (comment about the sequence data) followed by one or more lines of a nucleotide or amino acid sequence.
$ cat pro-refseq.fasta
>YP_009724390.1 |surface glycoprotein [Severe acute respiratory syndrome coronavirus 2]
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFS
NVTWFHAIHVSGTNGTKRFDNPVLPFNDGVYFASTEKSNIIRGWIFGTTLDSKTQSLLIV
...
QELGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDD
SEPVLKGVKLHYT
BioSeq utilites automatically remove the header and newlines to form a single tight line of nucleobases and amino acid codes.
You may want to create a raw data file by removing the header and newlines for easy handling. The following Linux command can be used to do that:
$ tail pro-refseq.fasta -n +2 $1 | tr -d '\n'
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV...
A shell script to call the above command exists:
$ rawfasta pro-refseq.fasta
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV...
The rawfasta script can be used between pipes:
$ cat pro-refseq.fasta | rawfasta | cut -c 3-5
VFL
ModsHunt can be used to compare sequence data.
The following command compares the spike proteins of the Wuhan and Australia viruses:
$ modshunt papro-refseq.fasta pro-au-200617.fasta
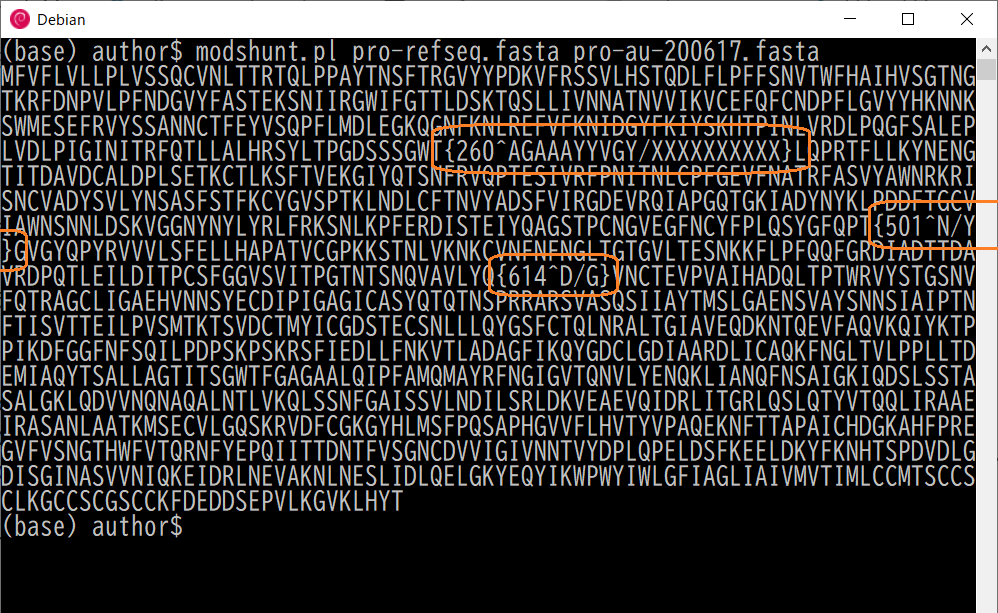
The comparison result shown is the source seuqence intermixed with differences:
| Difference | Description | Remark |
|---|---|---|
| {260^AGAAAYYVGY/XXXXXXXXXX} | Ten amino acides are deleted | |
| {501^N/Y} | The 501'th amino acid changed from N to Y | N501Y |
| {614^D/G} | The 614'th amino acid changed from D to G | D614Y |
You can show both sequences in parallel using the -fd format option:
$ modshunt -fd pro-refseq.fasta pro-au-200617.fasta
1
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV
......................................................................
1
~~~
260 270
NLVRDLPQGFSALEPLVDLPIGINITRFQTLLALHRSYLTPGDSSSGWTAGAAAYYVGYLQPRTFLLKYN
.................................................XXXXXXXXXX...........
260 270
~~~
502
PLQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVCGPKKSTNLVKNKCVNFNFNGLTGTGVLTESNKKFL
..........Y...........................................................
502
~~~
KNLNESLIDLQELGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDD
......................................................................
SEPVLKGVKLHYT
.............
You can output the difference data suitable for input to another program, such as a viewer, in a tab-separated format (-ft) or in Json format (-fj): In addition, -fx format option can be used to show in tab-separated format but with shorter strings for debugging purpose.
The next example compares the spike protein portion cut from the entire nucleotide sequences from Wuhan and Australia with -fx option:
$ cut -c 21563-25384 nuc-refseq.fasta > cut-refseq.data
$ cut -c 21525-25346 nuc-au-au-200617.fasta > cut-au-200617.data
$ modshunt -fx cut-refseq.data cut-au-200617.data
sync 1+777 1+777 "ATGTTTGTTTTTCTT..." "ATGTTTGTTTTTCTT..."
diff 778+28 778+28 "GCTGGTGCTGCAGCT..." "NNNNNNNNNNNNNNN..."
sync 806+695 806+695 "ATCTTCAACCTAGGA..." "ATCTTCAACCTAGGA..."
diff 1501+1 1501+1 "A" "T"
sync 1502+339 1502+339 "ATGGTGTTGGTTACC..." "ATGGTGTTGGTTACC..."
diff 1841+1 1841+1 "A" "G"
sync 1842+1981 1842+1981 "TGTTAACTGCACAGA..." "TGTTAACTGCACAGA..."
See a summary of the differences below. You see the differences correspond to the amino acid variations seen above before:
| Position | Variation in Nucleotides | Corresponding Variation in Amino Acids |
|---|---|---|
| 778'th character | 28 nucleobases are deleted | 10 amino acids are deleted |
| 1501'th character | Nucleobase A changed to T | N501Y (AAT>TAT) |
| 1841'th character | Nucleobase A changed to G | D614Y (GAT>GGT) |
You see counts of deleted nucleobases and amino acids do not match.
At the end, the same -fx option is used to compare entire genomes of coronavirus:
$ modshunt.pl -fx nuc-refseq.fasta nuc-au-200617.fasta
del 1+38 1+0 "ATTAAAGGTTTATAC..." ""
sync 39+202 1+202 "ACTTTCGATCTCTTG..." "ACTTTCGATCTCTTG..."
diff 241+1 203+1 "C" "T"
sync 242+2795 204+2795 "GTCCGGGTGTGACCG..." "GTCCGGGTGTGACCG..."
diff 3037+1 2999+1 "C" "T"
sync 3038+11370 3000+11370 "TACCCTCCAGATGAG..." "TACCCTCCAGATGAG..."
diff 14408+1 14370+1 "C" "T"
sync 14409+7931 14371+7931 "TACAAGTTTTGGACC..." "TACAAGTTTTGGACC..."
diff 22340+28 22302+28 "GCTGGTGCTGCAGCT..." "NNNNNNNNNNNNNNN..."
sync 22368+695 22330+695 "ATCTTCAACCTAGGA..." "ATCTTCAACCTAGGA..."
diff 23063+1 23025+1 "A" "T"
sync 23064+339 23026+339 "ATGGTGTTGGTTACC..." "ATGGTGTTGGTTACC..."
diff 23403+1 23365+1 "A" "G"
sync 23404+3580 23366+3580 "TGTTAACTGCACAGA..." "TGTTAACTGCACAGA..."
diff 26984+1 26946+1 "C" "T"
sync 26985+1211 26947+1211 "CATCTAGGACGCTGT..." "CATCTAGGACGCTGT..."
diff 28196+1 28158+1 "T" "C"
sync 28197+684 28159+684 "TGTTCGTTCTATGAA..." "TGTTCGTTCTATGAA..."
diff 28881+3 28843+3 "GGG" "AAC"
sync 28884+968 28846+968 "GAACTTCTCCTGCTA..." "GAACTTCTCCTGCTA..."
del 29852+52 29814+0 "AGCTTCTTAGGAGAA..." ""
NucleoAmino takes a part or whole of a nucleotide sequence, assumes the data as codes that encode a protein, and converts the data to an amino acid sequence.
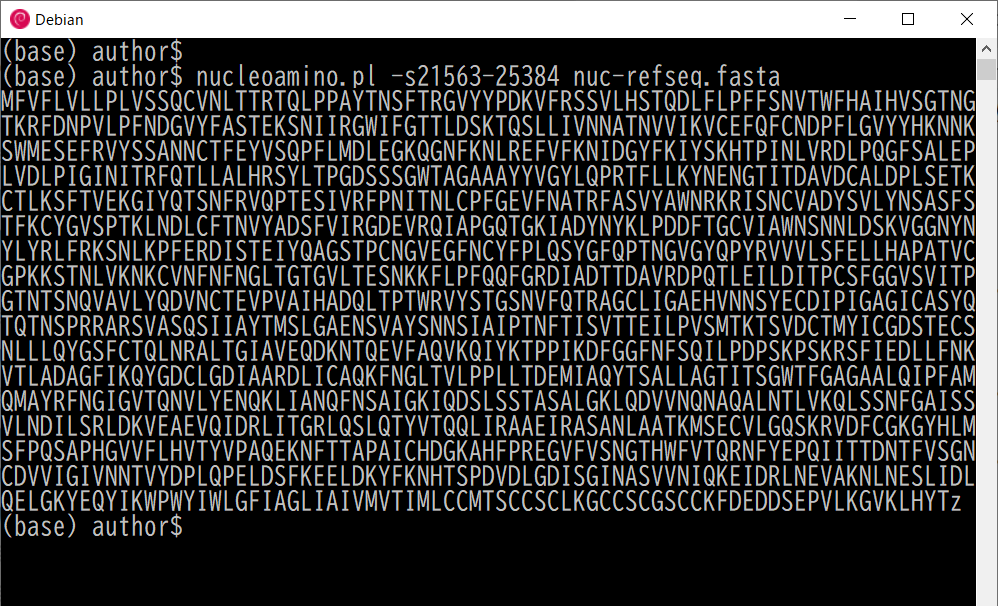
The next command converts the spike protein portion of the nucleotide sequence of the Wuhan virus to an amino acid sequence. The conversion range can be specified with -s option:
$ nucleoamino.pl -s21563-25384 nuc-refseq.fasta
You cannot simply compare two sets of genomes (DNA or RNA) or amino acid sequence even if they came from the same species because one or more nucleobases and amino acids can disappear or be added.
NCBI data are adjusted based on the Wuhan virus (RefSeq). Otherwise you need some measure to determine the corresponding parts by, for example, using a pattern of a portion that is relatively constant among many variants.
MotifGrep allows you to find a part that matches a pattern of a nucleotide or amino acid sequence. Such a pattern is called a motif (sequence motif). Here are some examples:
ATG matches against 'ATG' as isAT[GC] matches 'ATG' or 'ATC' where [..] means any character in the listAT{GC} matches 'ATT' or 'ATA' where {..} means any character not on the listI show you an example where MotifGrep is used to find a variation so called 'N501Y' within a spike protein's amino acid sequence.
In a research article that compares the first SARS and the current SARS-2 viruses, a comparison chart of the spike protein's amino acid sequences shows not-very-fast-changing parts in red:
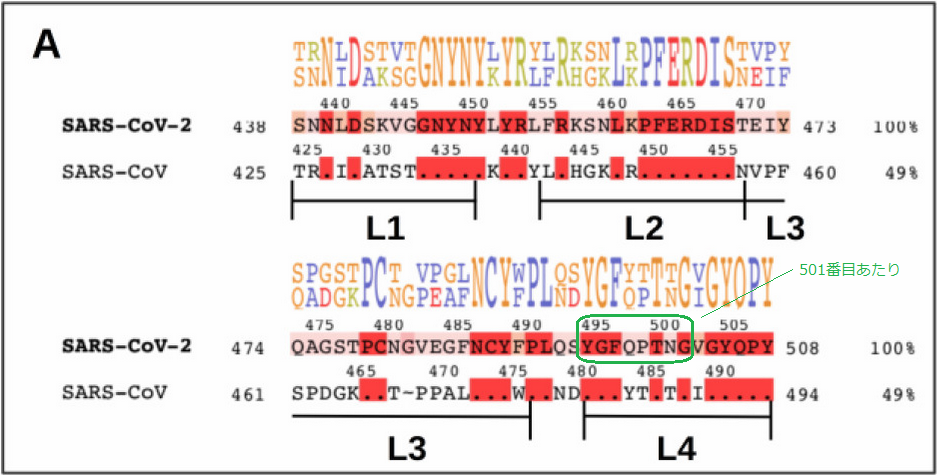
Is the Rigidity of SARS-CoV-2 Spike Receptor-Binding Motif the Hallmark for Its Enhanced Infectivity? Insights from All-Atom Simulations
(Angelo Spinello, Andrea Saltalamacchia, and Alessandra Magistrato)
pubs.acs.org/doi/10.1021/acs.jpclett.0c01148#
I used a motif pattern (YGFxxTx) appearing just before the N501Y variation (501'th amino acid) and used the pattern to find the corresponding parts of the data I have:
$ motifgrep YGFxxTx pro-refseq.fasta
495:YGFQPTN
$ motifgrep YGFxxTx pro-au-200617.fasta
495:YGFQPTY
You see the 501'th amio acid changed from N to Y.
MotifGrep can convert an amino acid pattern to nucleotide pattern in order to search nucleobases as a protein coding region:
$ motifgrep -n YGFxxTx nuc-refseq.fasta
23045:TATGGTTTCCAACCCACTAAT
$ motifgrep -n YGFxxTx nuc-au-200617.fasta
23007:TATGGTTTCCAACCCACTTAT
You see the nucleotide sequences corresponding to the 501'th amino acid seen before changed from AAT to TAT.
Off course you can simply search a nucleotide sequence by a nucleotide pattern.
$ motifgrep TATGGTTTCCAACCCACTAAT nuc-refseq.fasta
23045:TATGGTTTCCAACCCACTAAT
$ motifgrep TATGGTTTCCAACCCACTTAT nuc-au-200617.fasta
23007:TATGGTTTCCAACCCACTTAT
This is the end of introduction of the BioSeq utilities which help decoding of bio-sequences such as nucleotide sequences in a nucleic acid (RNA/DNA) or amino acid sequences of a protein.
Feel free to contact us for questions, problem reports, improvement requests and cusomization requests.
For your curiosity, here are ModsHunt's execution times measured with the first and revised versions of the code:
| File | Size | 2021/02/09 | 2021/02/19 | Remark |
|---|---|---|---|---|
| pro-xxxx.fasta | 1383 | Less than one second | - | Amino acid sequence of the spike protein |
| cut-xxxx.data | 3823 | Ten seconds | Five seconds | nucleotide sequence for the spike protein |
| nuc-xxxx.fasta | 30500 | Eleven minutes | Seven minutes | Entire Genome |
Test execution environment:
It took eleven minutes to compare entire coronavirus genome with the first version (02/09). It was reduced to seven minutes by removing unnecessary code without adding concurrency (02/19). There are some places available for concurrency. I am working on it.
Written: 2021/02/08
Updated: 2021/02/20