Home > BioSeq Utilities | English
BioSeqユーティリティは、核酸(RNA/DNA)の塩基配列やタンパク質を構成するアミノ酸配列などの生物情報(biological sequence)を解読するPerlスクリプトの集まりです。 以下では塩基またはアミノ酸の配列データをあわせて「バイオ配列」と呼びます。
生物情報の交換形式はたいていテキストファイルなので、テキスト処理に優れたUNIX(Linux)のコマンドラインを使うとたやすく取り扱うことができます。 BioSeqユーティリティはLinuxのコマンドラインツールを補い、バイオシーケンス特有の解読を手助けするツール群です。
Kobu.Comは長年にわたりテキスト処理(検索・加工・形式変換)に携わっており、特に大量文書のバッチ処理が得意です。 生物の情報処理(bioinformatics)は医療や生物の知識とコンピュータの知識の両方が求められます。 医療分野や生物研究に携わる方で計算が不如意という方はご支援可能ですのでご相談ください。 検査データ解析の準備、情報登録の準備などで必要とされるデータ処理のお手伝いが可能です。 バイオ配列の解読・加工・形式変換などを行うツールの設計、作成、カスタマイズなど可能ですのでご相談ください。
現在次の三つのユーティリティがあります(作った順)。
| ユーティリティ | 名前 | 説明 |
|---|---|---|
| motifgrep | パターン照合 | バイオ配列に出現するモチーフ(motif)パターンを検索 |
| nucleamino | 塩基とアミノ酸の変換 | 核酸の塩基配列をタンパク質のアミノ酸配列に変換 |
| modshunt | 配列の差分 | ふたつのバイオ配列を比較し差異を表示 |
BioSeqユーティリティはGitHubで公開しています。 セットアップおよび使用方法はGitHubのREADME.mdをごらんください(英語)。
以下、例を使いながらBioSeqユーティリティを紹介します。
なお、お時間があれば、先に下記の関連記事をごらんいただくとわかりやすいかと思います。 Linuxのコマンドラインでコロナウイルス(SARS-CoV-2)のRNA塩基配列や突起蛋白(S Protein)のアミノ酸配列を解読した体験記事です。
本題に入る前にユーティリティの使用例で取り扱うテストデータについて説明します。 米国の国立衛生研究所(NIH)が中心になって運営している生物情報データベース(NCBI)からコロナウイルス(SARS-CoV-2)の核酸(RNA)の塩基配列および突起蛋白(S Protein)のアミノ酸配列をダウンロードして使用します。
二種類のウイルスのデータを取り扱います。 ひとつは武漢で2019年12月に採取されたもので、変異株比較の基準となるウイルスデータ(RefSeq)とされているものです。 もうひとつは2020年6月に豪州で採取された変異株で、感染力が高まった要因と疑われる「N501Y」の変異を含みます。 両者ともRNAの塩基配列と突起蛋白のアミノ酸配列の二組のデータを取得しました(2021/01取得)。
| Virus | Sequence Type | Accession | File | S Protein Range |
|---|---|---|---|---|
| RefSeq | Nucleotide | NC_045512.2 | nuc-refseq.fasta | 21563..25384 |
| RefSeq | Surface Glycoprotein | YP_009724390.1 | pro-refseq.fasta | |
| Au N501Y | Nucleotide | MT745734.1 | nuc-au-200617.fasta | 21525..25346 |
| Au N501Y | Surface Glycoprotein | QLG76817.1 | pro-au-200617.fasta |
NCBIサイトにて登録番号(accession)を指定すれば同じファイルをダウンロードできます。
NCBIなどオンラインの生物情報データベースではFASTA形式が多く使われています。 FASTA形式のファイルは、'>'で始まるヘッダ行(配列データの由来)の後ろに塩基配列またはアミノ酸配列が続きます(一行または複数行)。
$ cat pro-refseq.fasta
>YP_009724390.1 |surface glycoprotein [Severe acute respiratory syndrome coronavirus 2]
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFS
NVTWFHAIHVSGTNGTKRFDNPVLPFNDGVYFASTEKSNIIRGWIFGTTLDSKTQSLLIV
...
QELGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDD
SEPVLKGVKLHYT
BioSeqユーティリティは自動的にヘッダと改行を取り除き塩基やアミノ酸コードがびっしりと並ぶ1行のテキストに直してから処理を開始します。
取り扱い上ヘッダや改行を削除したファイルを作成したい場合もあります。 それには次のようなLinuxコマンドで行えます。
$ tail pro-refseq.fasta -n +2 $1 | tr -d '\n'
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV...
上記のコマンドを呼び出すシェルスクリプトも用意してあります。
$ rawfasta pro-refseq.fasta
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV...
Rawfastaスクリプトはパイプの間に挟んでも使えます。
$ cat pro-refseq.fasta | rawfasta | cut -c 3-5
VFL
配列データの比較はModsHuntで行えます。
武漢と豪州のウイルスの突起蛋白を比較してみます。
$ modshunt papro-refseq.fasta pro-au-200617.fasta
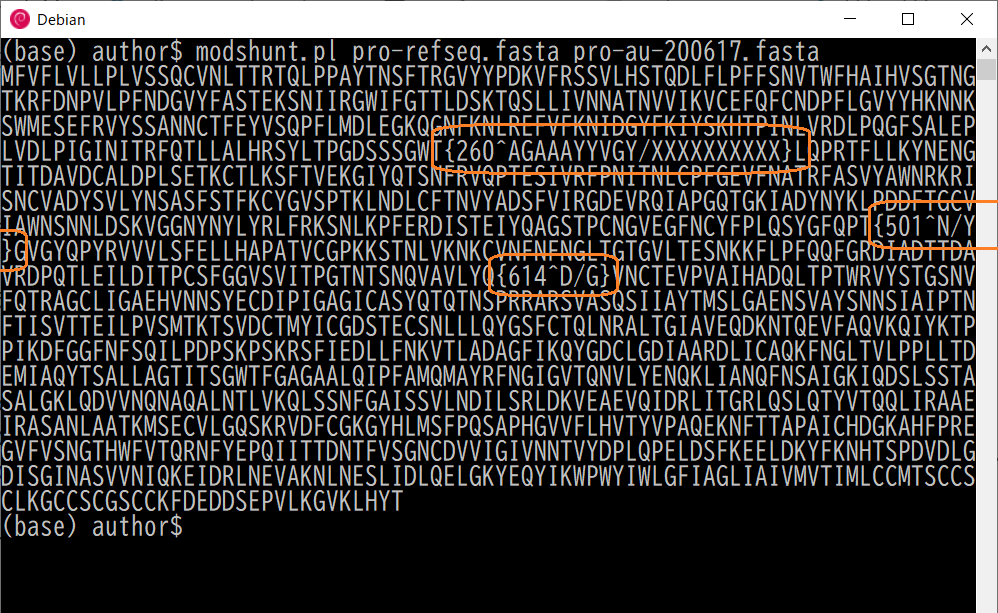
比較結果は比較元の配列の並びに差異部分を埋め込んだかたちで表示されます。
| 差異 | 説明 | 備考 |
|---|---|---|
| {260^AGAAAYYVGY/XXXXXXXXXX} | 10個のアミノ酸が削除された | |
| {501^N/Y} | 501番目のアミノ酸がNからYに変わった | N501Y |
| {614^D/G} | 614番目のアミノ酸がDからGに変わった | D614Y |
形式オプション-fdを指定して両者併記で表示も可能です。
$ modshunt -fd pro-refseq.fasta pro-au-200617.fasta
1
MFVFLVLLPLVSSQCVNLTTRTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFFSNVTWFHAIHV
......................................................................
1
~~~
260 270
NLVRDLPQGFSALEPLVDLPIGINITRFQTLLALHRSYLTPGDSSSGWTAGAAAYYVGYLQPRTFLLKYN
.................................................XXXXXXXXXX...........
260 270
~~~
502
PLQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVCGPKKSTNLVKNKCVNFNFNGLTGTGVLTESNKKFL
..........Y...........................................................
502
~~~
KNLNESLIDLQELGKYEQYIKWPWYIWLGFIAGLIAIVMVTIMLCCMTSCCSCLKGCCSCGSCCKFDEDD
......................................................................
SEPVLKGVKLHYT
.............
このほか、ビューアなど他のプログラムへの入力とするためタブ区切りテキスト(-ft)またはJson(-fj)で差分情報を出力することも可能です。 加えて基本はタブ区切りだがデバッグ用にシーケンスを省略して表示する-fxオプションもあります。
武漢と豪州のウイルスの全塩基配列から突起蛋白部分を切り出して-fxオプションで比較してみます。
$ cut -c 21563-25384 nuc-refseq.fasta > cut-refseq.data
$ cut -c 21525-25346 nuc-au-au-200617.fasta > cut-au-200617.data
$ modshunt -fx cut-refseq.data cut-au-200617.data
sync 1+777 1+777 "ATGTTTGTTTTTCTT..." "ATGTTTGTTTTTCTT..."
diff 778+28 778+28 "GCTGGTGCTGCAGCT..." "NNNNNNNNNNNNNNN..."
sync 806+695 806+695 "ATCTTCAACCTAGGA..." "ATCTTCAACCTAGGA..."
diff 1501+1 1501+1 "A" "T"
sync 1502+339 1502+339 "ATGGTGTTGGTTACC..." "ATGGTGTTGGTTACC..."
diff 1841+1 1841+1 "A" "G"
sync 1842+1981 1842+1981 "TGTTAACTGCACAGA..." "TGTTAACTGCACAGA..."
差異部分をまとめると次のとおりです。 先に見たアミノ酸配列の変化に対応していることがわかります。
| 位置 | 塩基の変異 | 対応するアミノ酸の変異 |
|---|---|---|
| 778文字目 | 28個の塩基の削除 | 10個のアミノ酸の削除 |
| 1501文字目 | 核酸塩基がAからTに変わった | N501Y (AAT→TAT) |
| 1841文字目 | 核酸塩基がAからGに変わった | D614Y (GAT→GGT) |
削除部分の塩基とアミノ酸の数が一致しません。
最後に同じく-fxオプションを使ってコロナウイルスのゲノム全体を比較してみます。
$ modshunt.pl -fx nuc-refseq.fasta nuc-au-200617.fasta
del 1+38 1+0 "ATTAAAGGTTTATAC..." ""
sync 39+202 1+202 "ACTTTCGATCTCTTG..." "ACTTTCGATCTCTTG..."
diff 241+1 203+1 "C" "T"
sync 242+2795 204+2795 "GTCCGGGTGTGACCG..." "GTCCGGGTGTGACCG..."
diff 3037+1 2999+1 "C" "T"
sync 3038+11370 3000+11370 "TACCCTCCAGATGAG..." "TACCCTCCAGATGAG..."
diff 14408+1 14370+1 "C" "T"
sync 14409+7931 14371+7931 "TACAAGTTTTGGACC..." "TACAAGTTTTGGACC..."
diff 22340+28 22302+28 "GCTGGTGCTGCAGCT..." "NNNNNNNNNNNNNNN..."
sync 22368+695 22330+695 "ATCTTCAACCTAGGA..." "ATCTTCAACCTAGGA..."
diff 23063+1 23025+1 "A" "T"
sync 23064+339 23026+339 "ATGGTGTTGGTTACC..." "ATGGTGTTGGTTACC..."
diff 23403+1 23365+1 "A" "G"
sync 23404+3580 23366+3580 "TGTTAACTGCACAGA..." "TGTTAACTGCACAGA..."
diff 26984+1 26946+1 "C" "T"
sync 26985+1211 26947+1211 "CATCTAGGACGCTGT..." "CATCTAGGACGCTGT..."
diff 28196+1 28158+1 "T" "C"
sync 28197+684 28159+684 "TGTTCGTTCTATGAA..." "TGTTCGTTCTATGAA..."
diff 28881+3 28843+3 "GGG" "AAC"
sync 28884+968 28846+968 "GAACTTCTCCTGCTA..." "GAACTTCTCCTGCTA..."
del 29852+52 29814+0 "AGCTTCTTAGGAGAA..." ""
NucleoAminoを使うと塩基配列の並びを蛋白質のコーディング部分とみなし、対応するアミノ酸配列に変換できます。 一個のアミノ酸は核酸塩基三つの並びで表されます。 たとえば、Asparagine(N)はAAUかAAC、Tyrosine(Y)はUAUかUACで表されます。
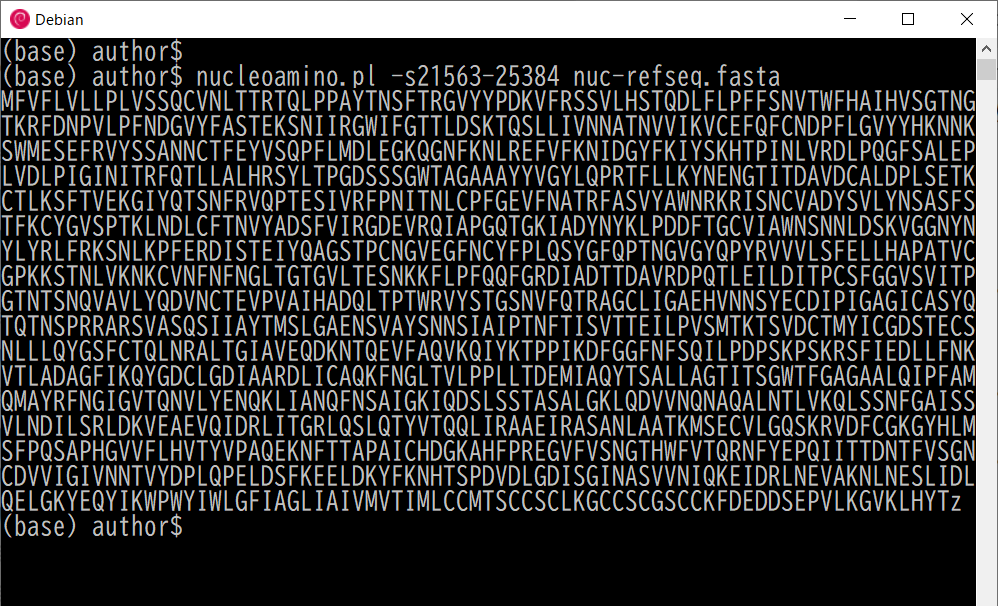
次のコマンドは武漢のウイルスの塩基配列の突起蛋白部分をアミノ酸配列に変換します。 変換部分の範囲指定は-sオプションで行います。
$ nucleoamino.pl -s21563-25384 nuc-refseq.fasta
同一種の生き物の異なる個体であっても、遺伝情報のDNAやRNA、アミノ酸配列を比較する場合、核酸塩基やアミノ酸が、いくつかなくなったり、増えたりするので、単純には比較できません。
NCBIのデータは武漢のウイルス(RefSeq)を基準として位置が調整されていて便利ですが、そうでない場合は、変異株を比較してあまり変わらない部分を見つけ、その箇所のパターンをたよりに対応する配列部分を見つける、などの手立てが必要です。
MotifGrepを使うと塩基配列やアミノ酸配列から特定のパターンを持つ箇所を探すことができます。 使用するパターンはモチーフ(sequence motif)と呼ばれるものです。 いくつか例を示します。
ATG → そのまま「ATG」と合致AT[GC] → [..]はいずれかの文字との合致を意味し「ATG」か「ATC」と合致。AT{GC} → {..}はそれ以外の文字との合致を意味し「ATT」か「ATA」と合致。x → どの文字とも合致MotifGrepの使用例として突起蛋白のアミノ酸配列から「N501Y」の変異箇所を見つける例を示します。
ある論文記事に一回目のSARSと今回のSARS-2ウイルスを比較したものがありました。 そのなかに突起蛋白のアミノ酸配列を比較した図があり、あまり変わらないとされる部分(赤い箇所)が示されてました。
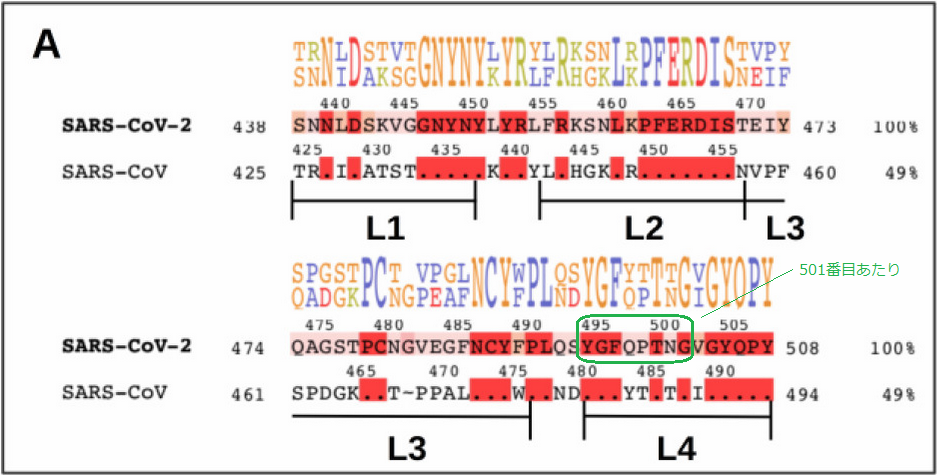
Is the Rigidity of SARS-CoV-2 Spike Receptor-Binding Motif the Hallmark for Its Enhanced Infectivity? Insights from All-Atom Simulations
(Angelo Spinello, Andrea Saltalamacchia, and Alessandra Magistrato)
pubs.acs.org/doi/10.1021/acs.jpclett.0c01148#
これをたよりに「N501Y」の変異箇所(501番目のアミノ酸)を直前のモチーフパターン(YGFxxTx)を使って見つけます。
$ motifgrep YGFxxTx pro-refseq.fasta
495:YGFQPTN
$ motifgrep YGFxxTx pro-au-200617.fasta
495:YGFQPTY
501番目がNからYに変化していることがわかります。
MotifGrepはアミノ酸配列のパターンを塩基配列のパターンに変換して、塩基配列をタンパク質のコード領域として検索することも可能です(-nオプションを追加)。
$ motifgrep -n YGFxxTx nuc-refseq.fasta
23045:TATGGTTTCCAACCCACTAAT
$ motifgrep -n YGFxxTx nuc-au-200617.fasta
23007:TATGGTTTCCAACCCACTTAT
先に見た501番めのアミノ酸に該当する元の塩基配列がAATからTATに変化していることがわかります。
もちろん、単純に塩基配列を塩基配列のパターンで検索することも可能です。
$ motifgrep TATGGTTTCCAACCCACTAAT nuc-refseq.fasta
23045:TATGGTTTCCAACCCACTAAT
$ motifgrep TATGGTTTCCAACCCACTTAT nuc-au-200617.fasta
23007:TATGGTTTCCAACCCACTTAT
核酸(DNA/RNA)の塩基配列やタンパク質のアミノ酸配列などバイオシーケンスの解読を手助けするBioSeqユーティリティの紹介は以上です。
ご不明点、不具合、改良案、カスタマイズのご要望などございましたら遠慮なくお尋ねください。
参考まで、初版および改訂版のコードで計測したModsHuntの実行速度を示します。
| ファイル | サイズ | 2021/02/09 | 2021/02/19 | 備考 |
|---|---|---|---|---|
| pro-xxxx.fasta | 1383 | 1秒以下 | - | 突起蛋白のアミノ酸配列 |
| cut-xxxx.data | 3823 | 10秒 | 5秒 | 塩基配列の突起蛋白部分 |
| nuc-xxxx.fasta | 30500 | 11分 | 7分 | 全塩基配列 |
実行環境は次のとおり。
初版のModsHuntでコロナウイルスの全ゲノムの差異を調べるのに11分かかりましたが(02/09)、 シーケンシャルのまま無駄な処理を省いたところ7分に短縮できました(02/19)。 並列化が可能な箇所があるのでさらに高速化が可能と思います。
Written: 2021/02/08
Updated: 2021/02/20